大规模实用化量子化学计算曙光显现,ByteDance Research开源工具集ByteQC
文章摘要
【关 键 词】 量子化学、GPU加速、开源工具、计算优化、材料模拟
大规模量子化学计算长期以来面临计算资源需求高、复杂度指数级增长的挑战,尤其是在材料、制药和催化等领域,精确模拟真实化学体系一直是一个难点。为了解决这一问题,字节跳动 ByteDance Research 团队开发并开源了 ByteQC,这是一个基于 GPU 加速的大规模量子化学计算工具集。该工具集通过利用现代 GPU 的强大算力,显著加速了多种标准量子化学算法,并结合量子嵌入方法,实现了在「黄金标准」精度下的大规模体系模拟。
在硬件层面,ByteQC 在现代 GPU 上高效实现了多种量子化学算法,包括平均场计算(如 Hartree-Fock 方法和密度泛函理论)以及后 Hartree-Fock 方法(如 Møller-Plesset 微扰理论、随机相位近似、耦合簇方法和量子蒙特卡洛方法)。这些算法的 GPU 化不仅提高了计算速度,还扩展了可计算的体系规模。在算法层面,ByteQC 引入了一种量子嵌入方法,能够在保持高精度的同时,显著提升计算效率。
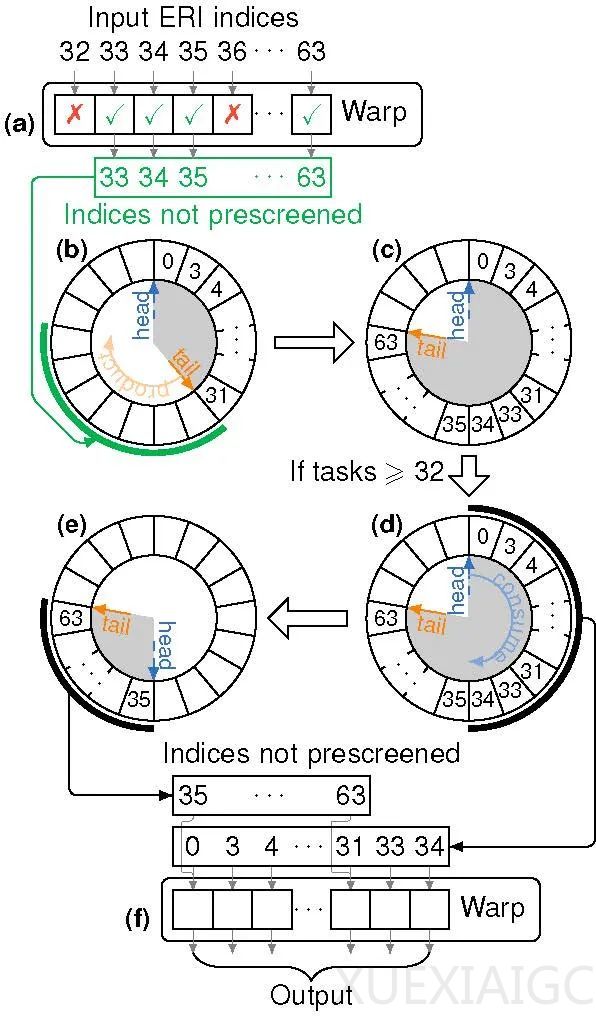
为了克服 GPU 显存有限和复杂逻辑难以高效实现的问题,ByteQC 在开发过程中采用了多种优化策略。首先,团队引入了 NVIDIA 提供的高效张量计算库 cuTENSR/cuTENSORMG,用于高效处理张量缩并操作,并在最小显存占用下完成计算。其次,团队通过动态 warp 特例化实现了高效的动态生产者-消费者模型,解决了周期性体系屏蔽计算中的复杂逻辑问题。此外,团队还优化了缓存使用,并通过 CUDA kernel 实现了原位操作,进一步减少了显存需求。
基准测试结果表明,ByteQC 在单 A100 GPU 上相比 100 核 CPU 实现了最高 60 倍的加速,大多数模块在多卡环境下也能达到线性加速。具体而言,ByteQC 能够处理的体系规模显著提升,例如耦合簇单、双激发(CCSD)可支持 1,610 轨道,带微扰三重激发(CCSD (T))可支持 1,380 轨道,二阶 Møller-Plesset 微扰理论(MP2)可支持 11,040 轨道,而开放边界条件下的平均场计算可支持 37,120 轨道,周期边界条件下的平均场计算甚至可支持超过 100,000 轨道。此外,结合量子嵌入功能,ByteQC 在 2,753 轨道的水团簇问题和 3,929 轨道的氮化硼表面水吸附问题上均实现了 CCSD (T) 水平的「黄金标准」精度计算。
ByteQC 的成功开发不仅解决了 GPU 在量子化学计算中的显存和复杂逻辑问题,还通过量子嵌入方法实现了更大规模体系的高精度计算。 这一工具集的开源有望推动量子化学领域的发展,为材料、制药和催化等领域的科学研究提供强有力的支持。
原文和模型
【原文链接】 阅读原文 [ 1213字 | 5分钟 ]
【原文作者】 机器之心
【摘要模型】 deepseek-v3
【摘要评分】 ★★★☆☆


相关文章



